Sequential Multiplex Editing with Hydropore Enables Universal Armored CAR-T Therapies
What Is a Universal CAR-T Cell?
Universal CAR-T cells, also known as “off-the-shelf” CAR-T cells, are designed to be used across different patients without customizing an individual’s T-cells1. Like conventional autologous CAR-T cell therapy, allogeneic universal CAR-Ts are engineered to recognize and destroy certain cancer cells. However, universal CAR-Ts have additional modifications to prevent them from attacking the patient’s (host’s) cells, known as graft-versus-host disease (GvHD). These modifications allow T-cells from a single healthy donor to treat multiple patients2.
“Off-the-shelf” CAR-Ts are desirable for several reasons. First, the availability would expand dramatically. Autologous therapies use a “made-to-order” process of collecting the patient’s blood, then isolating, engineering, and expanding the CAR-Ts before re-infusing them back into the patient. Universal CAR-Ts can be manufactured ahead of time and stored for future use3. This not only increases access to lifesaving treatments but also reduces manufacturing costs as the therapies are produced on a large scale.
Lastly, universal CAR-Ts potentially impact a broader patient pool. Many candidates for adoptive immunotherapy have weak or damaged immune systems either from their disease or previous treatment (i.e., chemotherapy and radiation)4. This limits the treatment’s therapeutic potential due to the challenges with unhealthy starting material. As universal CAR-Ts always start with healthy donors, the state of the patient’s immune system doesn’t impact the efficacy of the cell therapy.
What Makes a CAR-T Cell Armored?
Armored CAR-T cells are equipped with additional features to help them survive the tumor microenvironment (TME) and proliferate. They can exert their anti-tumor effects even in hostile conditions. CAR-T cells can be armored in different ways, but the most common include cytokine secretion, immunosuppression resistance, multiple target antigens, TME modulation, and enhanced persistence5.
Here, we armored our CAR-T cells against immunosuppression by knocking out the CD5 gene. CD5 functions as a negative regulator of T-cell receptor (TCR) signaling. By knocking out CD5, CAR-T cells experience a stronger, more sustained activation signal when they engage tumor cells. This leads to a more robust anti-tumor effect with greater proliferation and increased cytotoxicity6.
What Is Multiplex Gene Editing?
Multiplex gene editing confers multiple genetic modifications at different loci within a cell’s genome. This is advantageous because it allows multiple functions to be added or deleted7. Here, we conferred three modifications. First, we knocked out beta-2-microglobulin (B2M), which effectively turns off the human leukocyte antigen (HLA), the cell surface marker that allows the immune system to recognize “self” versus “non-self”8. This is the first step in making a CAR-T cell universal.
Next, we knocked-in an anti-CD19 CAR into the TRAC-1 locus. This modification served two purposes. First, by silencing the endogenous TCR through TRAC-1 disruption, the cell loses any alloreactivity and potential to cause GvHD. Second, by knocking-in an anti-CD19 CAR to the TRAC-1 locus, the cell is now equipped with a synthetic receptor that promotes T-cell activation upon engaging with its antigen. In this case, the antigen is CD19, a surface protein that is over-expressed in B cell malignancies.
Finally, we armored the cell against immunosuppression by knocking out CD5.
What Are the Challenges of Multiplex Editing?
One of the primary challenges in multiplex gene editing within T-cell therapies is the difficulty in achieving high yields of homogeneously edited T-cells. Each additional gene edit decreases the proportion of cells successfully modified. Plus, the transfection process itself is associated with significant cytotoxicity9. As a result, the culture often becomes highly heterogeneous, with only a small subset of cells displaying all of the desired modifications.
This issue is exacerbated when attempting to deliver larger genetic payloads, such as a chimeric antigen receptor (CAR). Standard approaches to multiplex editing typically involve multiple guide RNAs (gRNAs) targeting distinct genomic loci concurrently. This often necessitates the construction of polycistronic expression cassettes via plasmid-based cloning, which are then incorporated into lentiviral vectors for stable, constitutive expression of the gRNAs10.
This strategy presents several limitations, including the high costs associated with lenti/retro- viral vector production and the potential for random genomic integration, which can lead to off-target effects and immunogenic responses11.
Furthermore, simultaneous induction of double-strand breaks (DSBs) across multiple sites can result in extensive genomic rearrangements, compromising the functionality of the edited cells12. This is particularly problematic when co-delivering distinct Cas9 ribonucleoprotein complexes. These challenges, especially those related to multiple gene cut sites, can be mitigated through the sequential delivery of the gene editing payloads, as demonstrated in this study.
Sequential Processing for Multiplex Editing
We compared the generation of multiplex-edited CAR-T cells made via hydroporation or nucleofection. In order to do this, PBMCs were thawed, and T-cells were isolated before being activated for 48 hours. On the day of transfection, activated T-cells were mixed with a B2M-targeting RNP and treated by hydroporation or nucleofection (Figure 1).
Cells were allowed to rest for 24 hours before being treated to a second round of CRISPR KO with a TRAC-1-targeting RNP. T-cells were then incubated with an AAV containing a CD19 CAR for 24 hours before a third round of KOs, this time with a CD5-targeting RNP. Cells were cultured for up to 10 days, with a portion of CAR-T cells treated for TCR depletion for use in a cytotoxicity assay, where they were co-cultured with luciferase-expressing NALM6 cells.

Figure 1. Overview of workflow for generation of CAR-Ts through RNP and AAV transfection, and how hydroporation is incorporated into the cell therapy workflow with improved cell numbers and viabilities for downstream analysis
Hydroporated T-cells Showed Better Recovery and Proliferation Capacity vs. Nucleofected T-cells
Our results show that while three days of consecutive nucleofection has a permanent negative impact on T-cell health, T-cells subjected to the same regimen of hydroporation are able to recover and maintain high proliferative capacity. After the third day of RNP delivery, T-cell viability for both hydroporated cells and electroporated cells was between 30 - 40% (Figure 2A).
However, hydroporated cell viability recovered to ~70% by day 5, while electroporated cells maintained low viability, hovering around 30%. Furthermore, over the next five days, hydroporated cells expanded 3.2 fold while maintaining viability above 70%. During that same period, nucleofected cell viability fell to 14%, and cell numbers dropped 4.8 fold (Figure 2A, B).
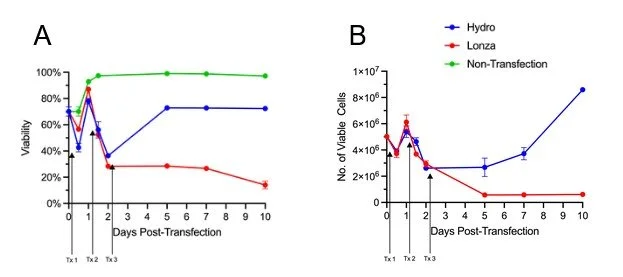
Figure 2. CAR-Ts transfected via hydroporation for three consecutive days showed greater viability and proliferation when compared to nucleofected cells. A) Viability comparison of T-cells transfected by hydroporation (Hydro; Blue) vs nucleofection (Lonza; Red) over ten days, with day of transfection indicated by arrows B) Proliferation of T-cells from day 0 (transfection) to day 10. All data points involve n = 3 biological replicates.
Multiplex-Edited Hydroporated CAR-T Cells Showed Greater Yields vs. Those Nucleofected Three Times
Nucleofection demonstrated higher editing efficiency compared to hydroporation for all target loci, ranging from 85% to 95% for single locus knockouts. Hydroporation resulted in 58% to 62% knockout efficiency, depending on the target locus. In regards to transgene insertion, electroporation achieved 52% CAR+ knock-in compared to Hydropore’s 40% CAR+ knock-in (Figure 3A).
The double editing efficiency (CAR+, B2M- or CAR+, CD5-) for electroporation and hydroporation, on average, was 50% and 25%, whereas triple editing efficiency (CAR+, B2M-, CD5-) for electroporation and hydroporation was 47% and 16% (Figure 3B).

Figure 3. CAR-Ts transfected via hydroporation showed better yields by Day 10 when compared to nucleofected cells. A) Frequency of single targeted gene edits when cells are transfected by hydroporation (Hydro; Blue) vs nucleofection (Lonza; Red). B) Showing the editing efficiency of CAR KI either as a single edit, a double edit, or a triple edit by staining for EGPR+ cells. C) Total CAR-T yield on day 10. All data points involve n = 3 biological replicates.
Despite electroporation having a higher editing efficiency in the triple-edited CAR-T cells, hydroporation yielded 4.5x universal armored CAR-T cells by day 10 due to superior cell viability and proliferation (Figure 3C).
Multiplex-Edited Hydroporated CAR-T Cells Performed As Well As Those Edited via Nucleofection
In regards to effector function, hydroporated and electroporated CAR-Ts performed similarly in a co-culture cytotoxicity assay. Target cell lysis ranged from 10% to 99% as the number of effector cells increased. Visually, the hydropore killing curve is superior for 1:8 to 1:32 effector to target ratios; however, there is no statistical significance (Figure 4).
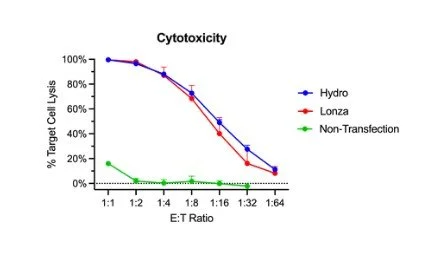
Figure 4. CAR-Ts modified through hydroporation showed similar levels of cytotoxicity. Bulk cytotoxicity assay based on treated T-cells at different ratios of effectors to target cells (CAR-T to NALM6).
Summary and Future Work
From previous work, we have shown the efficacy of hydroporation in the generation of functional CAR+ T-cells, with higher yields and similar functionality to those generated by conventional electroporation13. Here, we show that hydroporation can be used to generate universal CAR+T cells through multiplex editing, which are as functional as those generated by nucleofection but with greater viability and cell number.
Additional experiments to validate are planned in collaboration with CellChorus. They will use their Time-lapse Imaging Microscopy in Nanowell Grids (TIMING) assay, as well as Next Generation Sequencing (NGS), to validate our multiplex edited CAR+ T-cells.
Hydroporation can be incorporated into the CAR-T cell therapy pipeline, providing a faster and more robust system for treating cancer patients.
References
11. Milone, M. C. & O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 32, 1529–1541 (2018).